





Main
Functional interactions among combinations of genetic elements underlie many natural and engineered phenotypes1,2,3, often involving higher-order (≥ 3-plex) combinations of coding4,5 or non-coding elements6,7,8,9. Experimentally testing higher-order combinations of genetic perturbations has been limited by throughput, with prior systematic analyses primarily performed in yeast10,11,12,13,14. In mammalian functional genomics, pooled screens15,16,17 using sequencing readouts have been limited to up to three genetic perturbations per cell when using RNA interference18 or CRISPR-Cas9 (ref. 19). Further multiplexing in Cas9-based pooled screening is challenging due to increasingly complex cloning schemes for large constructs encoding multiple guides expressed from separate promoters and length-dependent high recombination frequencies of lentiviral guide libraries20,21,22. Conceptually, it also remains unclear how to tractably survey the potentially vast combinatorial spaces for ≥3-plex perturbations.
Cas12a, a member of the type V CRISPR-Cas family, has been proposed as an alternative to Cas9 for genetic perturbations due to enhanced multiplexing capabilities. Cas12a harbors RNase activity, separable from its DNase activity, that can process a compact primary transcript expressed from a single promoter into multiple CRISPR RNAs (crRNAs)23,24. An array of multiple Cas12a crRNAs, each composed of a 19-nt direct repeat and a 19-to 23-nt spacer, can be encoded by a chemically synthesized oligo for single-step cloning into an expression vector25,26,27,28,29. Cas12a has been engineered for mammalian cell applications using its DNase activity to disrupt coding gene function using single or multiplexed crRNA constructs in individual well-based assays24,25,26,27,30,31 and in pooled sequencing screens28,29,31,32,33,34,35. However, extended multiplexing with fully DNase-competent Cas12a is expected to be constrained by genotoxicity from double-stranded DNA breaks in many biological contexts28,36,37,38,39,40,41. In principle, avoiding genotoxicity can be achieved by using DNase-dead Cas fusion proteins to control chromatin state and transcription, such as by DNase-dead Cas9 (dCas9)-based CRISPR interference (CRISPRi) or CRISPR activation (CRISPRa)42,43,44. Moreover, CRISPRi is more efficient than DNA cutting at perturbing enhancers in pooled screens45,46,47, likely due to CRISPRi’s larger genomic window of activity via formation of repressive chromatin48. Thus, a DNase-dead Cas12a (dCas12a) functional genomics platform for multisite CRISPRi targeting would be highly desirable for testing the combinatorial functions of coding and non-coding genetic elements. However, no dCas12a-based pooled CRISPRi screening platform has been reported. Several studies have used dCas12a fusion proteins for CRISPRi in human cells in individual well-based assays, reporting either successful27,49,50,51 or unsuccessful52 repression of target genes. These dCas12a CRISPRi studies delivered crRNA plasmids by transient transfection rather than lentiviral transduction. Transient plasmid transfections express synthetic components at 10- to 1,000-fold higher than single-copy lentiviral integration of crRNA constructs, which is required in pooled screens to attribute cellular phenotypes to unique crRNA constructs by high-throughput sequencing15,16. Whether prior dCas12a CRISPRi constructs are sufficiently potent for pooled screens remains unclear.
In this study, we show that existing dCas12a CRISPRi fusion constructs function poorly when used with limiting doses of lentivirally delivered components, thus precluding their application in pooled screens. We engineered an Acidaminococcus Cas12a (AsCas12a) variant that incorporates a key mutation, R1226A, which enhances stability of the ribonucleoprotein–DNA complex in the form of a nicked DNA intermediate in vitro53,54. We show that in human cells, multiAsCas12a-KRAB fusion substantially improves CRISPRi activity in the setting of lentivirally delivered crRNA constructs, enabling use of 6-plex crRNA arrays in high-throughput pooled screens and up to 10-plex crRNA arrays in well-based assays. We use this combinatorial CRISPRi platform to efficiently discover enhancer elements and to test higher-order combinatorial perturbations of cis-regulatory elements. These results instantiate a group testing framework that enables efficient searches of potentially large combinatorial spaces of chromatin perturbations.
Results
Lentivirally delivered CRISPRi by dAsCas12a fusion proteins is hypoactive
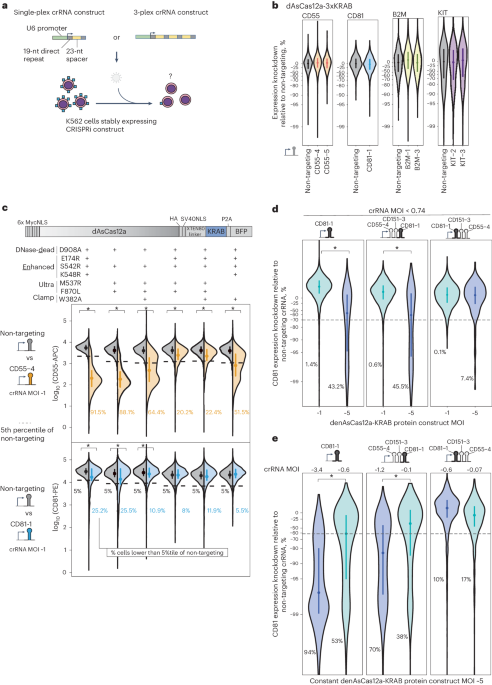
We focused on building a CRISPRi functional genomics platform using AsCas12a, the only Cas12a ortholog with demonstrated success in pooled screens in mammalian cells28,29,31,32,33,34,55,56. A previous study reported using dAsCas12a for CRISPRi by plasmid transient transfection delivery of dAsCas12a-3xKRAB protein (harboring the E993A DNase-dead mutation) and crRNA in HEK 293T cells27. To test this construct in the setting of lentivirally delivered crRNA, we introduced dAsCas12a-3xKRAB by piggyBac transposition in K562 cells, followed by lentiviral transduction of single crRNA constructs targeting canonical (TTTV) or non-canonical30 protospacer adjacent motifs (PAMs) proximal to transcriptional start sites of four genes encoding for cell surface proteins, three of which (CD55, CD81 and B2M) have been successfully knocked down by dCas9-KRAB CRISPRi (Fig. 1a)57. Throughout this study we encoded crRNAs in a CROP-seq58 lentiviral vector previously optimized for pooled screens using DNase-active AsCas12a (ref. 29). We observed no expression change in any of the targeted genes (Fig. 1b and Supplementary Figs. 1 and 2). We confirmed the expression of dAsCas12a-3xKRAB by western blot (Supplementary Fig. 3) and by flow cytometry monitoring of the in-frame P2A-BFP (Supplementary Fig. 4a). We also observed this lack of CRISPRi activity for dAsCas12a-3xKRAB using lentivirally transduced crRNAs in C4-2B prostate cancer cells (Supplementary Fig. 4b). In contrast, transient co-transfection of dAsCas12a-3xKRAB and CD55-targeting crRNA plasmids shows modest CRISPRi knockdown in HEK 293T cells (Supplementary Fig. 5), consistent with prior work27. These findings indicate that the requirements for CRISPRi activity using dAsCas12a-3xKRAB with lentiviral crRNA constructs are distinct from those of plasmid transient transfection in HEK 293T cells27.
Fig. 1: dAsCas12a-KRAB variants are dose-limited and weak in CRISPRi activity when using lentivirally delivered crRNAs, despite incorporating state-of-the-art optimizations.
a, Schematic for assaying CRISPRi activity of AsCas12a constructs using lentivirally transduced single-plex or 3-plex crRNAs targeting cell surface marker genes assayed by antibody staining and flow cytometry. b, K562 cells constitutively expressing dAsCas12a-3xKRAB27 were lentivirally transduced with the indicated single crRNAs and assayed by flow cytometry 6 days after crRNA transduction. One of two biological replicates is shown; second replicate is shown in Supplementary Fig. 2. c, A panel of AsCas12a variants harboring combinations of mutations are tested using crCD55-4 and crCD81-1 using the fusion protein domain architecture shown. Both AsCas12a fusion protein and crRNA constructs are delivered by lentiviral transduction. D908A is a mutation in the RuvC catalytic triad that renders Cas12a DNase inactive24,54. Other mutations are described in detail in the main text. Shown are single-cell distributions of target gene expression assayed by flow cytometry 6 days after crRNA transduction for one of three independent replicates. Additional replicates and results for additional crRNA constructs (up to 3-plex crRNA constructs) are summarized in Supplementary Fig. 6a–c. d, Analysis of CD81 knockdown in cells lentivirally transduced with denAsCas12a-KRAB protein construct at multiplicity of infection (MOI) ~1 versus MOI ~5 while maintaining constant crRNA MOI ( 200 cells per replicate). c–e, Asterisks indicate P
>>> Read full article>>>
Copyright for syndicated content belongs to the linked Source : Nature.com – https://www.nature.com/articles/s41587-024-02224-0


{kind=link}